
Molecular Modeling & Drug Discovery
3. Folding:
Domains & Folds
Domains
Condensed protein domains that are structurally (and typically functionally) separate.
a region of the protein that would keep its distinctive structure even if it were cut off from the rest of the protein.
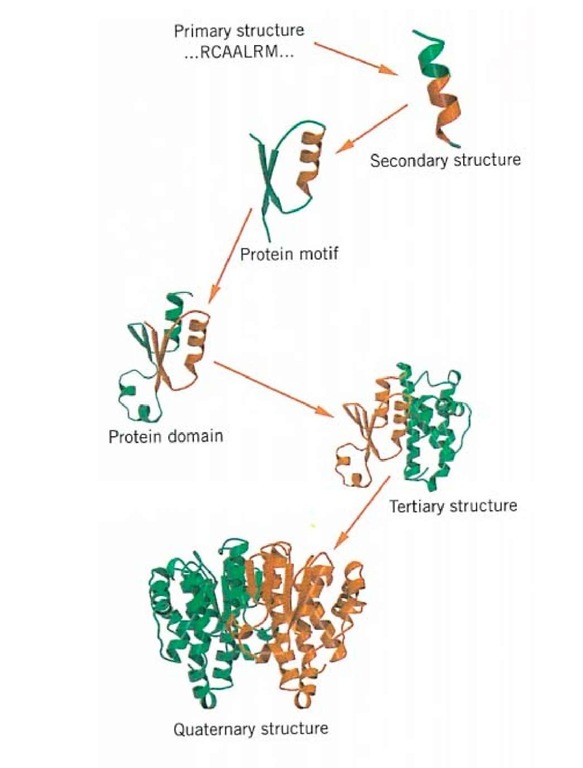
Figure 1. Proteins domains and motifs form an integral part of protein tertiary structure.
Folds
They are little substructures that don't necessary have independent structural design and merely have a few secondary structural components.
The same specific motifs can be found in numerous distinct protein architectures.
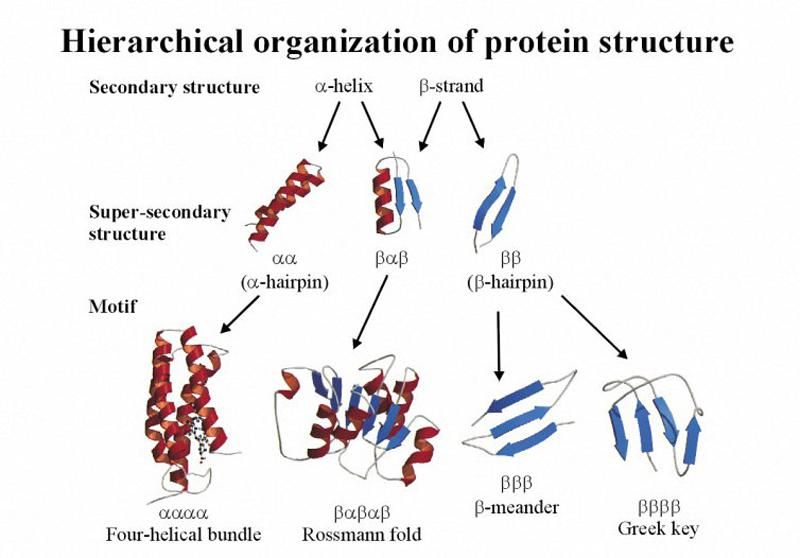
Figure 2. A motif is a brief, conserved sequence pattern connected to several DNA or protein functions. It frequently has a specific structural place performing a specific function attached to it.
Several motifs can unite to form certain domains.
They frequently have a functional importance and, in these circumstances, represent a minimal functional unit inside a protein.
3D-Figure 1. PDB structure of 2BK8 is a M1 domain from titin with sequence length of 97 and 1 chain (CLICK me for more info!!).
Tertiary structure molecular interactions
The protein fold is determined by the interactions between the structural components and the side chains that make up each component.
In addition to interactions inside the protein itself, intermolecular interactions with water (solvent) must be taken into account.
3D-Figure 2. 3D interactive map of structure of 2BK8 with red dots indicative of water molecules and main domain indicated by default as dark green.
Hydrophobic effect
While charged and polar residues make up the protein's surface and can interact with polar water molecules and solvated ions, hydrophobic residues are crammed into the protein's center, far from the solvent.
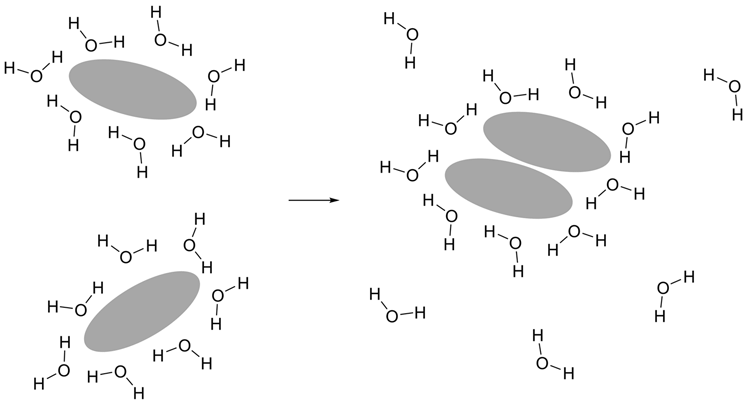
Figure 3. The hydrophobic effect is the main factor behind protein folding. Hydrophobic molecules have a propensity to distance themselves from interaction with water.
Hydrogen bonds
Hydrophobic residues can naturally fit into the protein's core, but their polar polypeptide backbone cannot.
The hydrogen bonding groups on the backbone must be satisfied in a way that effectively neutralizes their polarity in order for it to participate in the hydrophobic core.
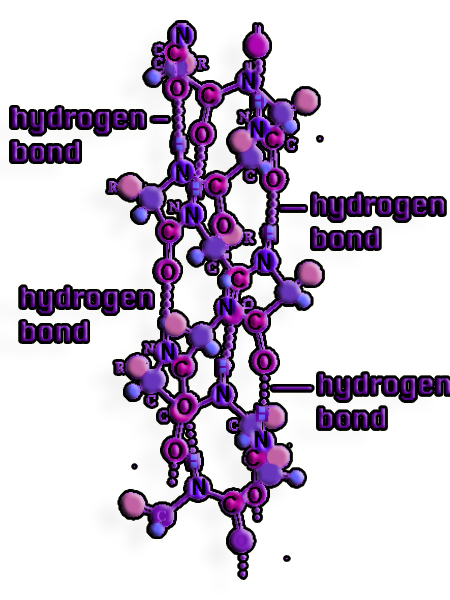
Figure 4. Hydrogen bonds arise between the backbone oxygens and amide hydrogens in the secondary structure of proteins. An alpha helix is created when the amino acid residues involved in hydrogen bonds are spaced regularly between positions I and I + 4.
Helices and sheets, ordered secondary structural components, offer this neutralization through consistent hydrogen bonding patterns.
The development of secondary structural components is essential for the hydrophobic core.
H bonds
Polar side chained residues can also take part in the hydrophobic core.
Similar limitations apply to them as to the polypeptide backbone: they need to interact in a way that balances their polarity.
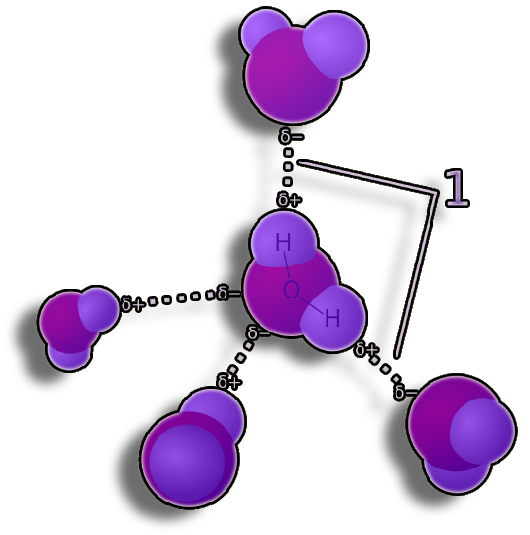
Figure 5. A hydrogen atom that is covalently joined to an electronegative atom (donor) interacts with another electronegative atom to form a hydrogen bond (acceptor). The stiffness of the protein structure and the selectivity of intermolecular interactions are both attributed to hydrogen bonds.
Polar residues that are buried in a polypeptide can create hydrogen bonds with other polar residues or with polypeptide backbone segments that aren't involved in a typical secondary structure.
Small spaces in some proteins allow buried polar residues to fulfill their hydrogen bonds with water molecules. These water molecules are essential to the structure of the protein and are totally isolated from the solvent (structural water).
Salt bridges
Charged residues can also exist inside the hydrophobic core, but they can be coupled with oppositely charged residues to make the net charge zero.
An ion pair or salt bridge is the name given to this interaction.
The anionic carboxylate
(RCOO) of either
aspartic acid or glutamic acid and the
cationic ammonium
(RNH3+) from
lysine or the guanidinium (RNHC(NH2)2+) of arginine most frequently form
the salt bridge.
The participation of other residues with ionizable side chains, such as
histidine, tyrosine, and serine, despite the fact that these are the
most frequent, depends on external circumstances that alter their pKas.
Another factor regarded as being crucial is the separation between the
residues taking part in the salt bridge.
The separation is under 400 pm or 4 armstrong.
More space between amino acids disqualifies them from building a salt
bridge. The pH at which a protein is placed is critical to its stability
since a protein contains multiple ionizable side chains of amino acids.
Covalent bonds
The thiol (— SH) groups of two cysteine residues
can form the disulphide bond (—S—S—).
In order to better stabilize the protein fold, this interaction is
present in proteins.
The bulk of these residues are not involved in a disulfide linkage,
hence cysteine residues are not always involved in disulfide bonds.
Two atoms share an electron pair equally in a covalent link.
Peptide (amide) and disulfide links between amino acids, as well as C-C, C-O, and C-N bonds within amino acids, are examples of significant covalent bonds.
Fold space
The term "fold space" refers to the possible protein folds.
The protein structure data that are now available indicate that fold space is actually rather limited (1,000-10,000).
Protein folds that evolved from a relatively small collection of related common ancestor proteins show divergent evolution of protein function.
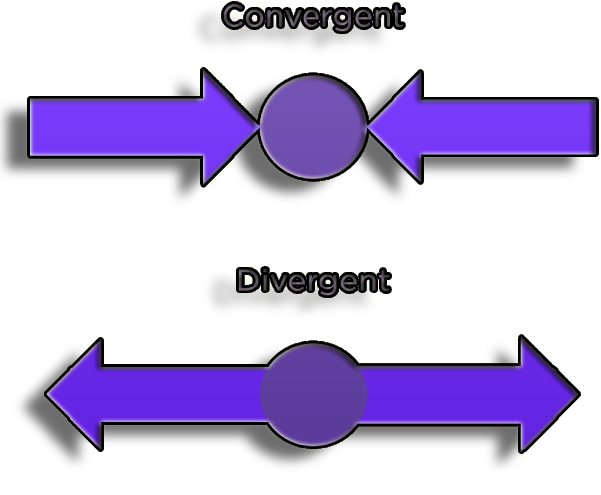
Figure 6. DNA and the amino acid sequences generated by the translation of structural genes into proteins converge.
Protein function and structure are not directly related. While protein function is influenced by protein structure, similar protein structures may not always have the same activities.
Protein structure has evolved convergently as a result of the biophysical preferences of particular folds, which have been independently produced in numerous instances.
In certain instances, proteins with identical folds have no sequence in common, indicating that they separately arrived at the same structure and did not have a common ancestor.
Folds are classified biochemically
Globular proteins are well represented in the PDB, in part because their structures are the simplest to identify experimentally.
They fold into compact forms with hydrophobic cores and polar surfaces because they exist in an aqueous environment.
Myoglobin and hemoglobin, the first two experimentally identified structures, are spherical.
3D-Figure 3. PDB structure of 1A6M is a oxy-myoglobinwith sequence length of 151 and 1 chain (CLICK me for more info!!).
Membrane proteins
The cell membrane is a hydrophobic environment where they are present.
A hydrophobic surface is necessary for the parts of the protein that are inside the cell membrane to be stable.
While some proteins have membrane-spanning or membrane-interacting domains, some proteins live almost exclusively within the cell membrane.
.jpeg)
Figure 7. Various types of membrane proteins have been depicted above.
The experimental determination of the structure of membrane proteins is more challenging than that of globular proteins. In the PDB, these proteins are not well represented.
More and more 3D structures have been solved in recent years. They employ the same secondary structural components as globular proteins and adhere to similar overall folding rules.
Fibrous proteins
Differ significantly from membrane proteins and globular proteins.
Frequently made up of simple, elongated fibers that are composed of repeating amino acid sequences.
Design repetition is ideal for structural tasks.
3D-Figure 4. PDB structure of 1QSU is a triple helical collagen peptide sequence length of 30 and 3 chains (CLICK me for more info!!).
Some fibrous proteins are made up of very lengthy sequences of the same kind of regular secondary structure. Others lack any discernible secondary structure or are composed of repeated or typical secondary structures.
Structural
classification of folds
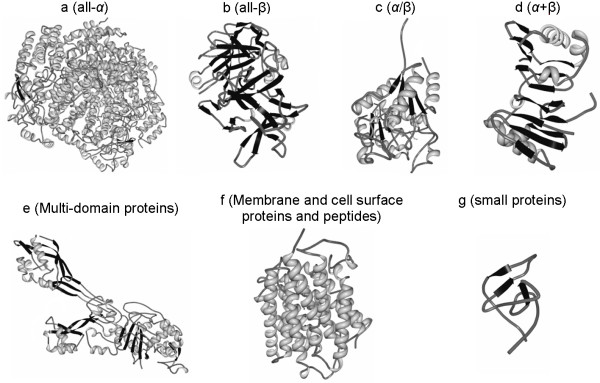
Figure 8. Various types of secondary classification of protein.
-
all α: based essentially exclusively on α-helical structure.
-
all β: based almost entirely on β-sheet.
-
α/β: Based on a combination of β-sheet and α-helix, commonly arranged as parallel β-strands joined by α-helices.
-
α + β: Distinct α-helix and β-sheet motifs that are not intertwined, as they are in a α-helix and β-sheet structure.
Quaternery structure
proteins that are made up of two or more independently folded polypeptides joined together (noncovalently) (multisubunit or multimeric proteins
identical subunits make up homomeric proteins (or protomers)
Different protomers make up a heteromeric protein.
The same kinds of interactions that are used to stabilize secondary and tertiary structures are also used to stabilize quaternary structures. The cores of globular proteins typically resemble the surface regions engaged in subunit interactions.
Tetramer consisting of two α or and two β subunits non-covalently bound, each made of 141 and 146 amino acid residues, respectively (α2β2).
3D-Figure 5. PDB structure of 1A3N is a human haemoglobin of sequence length of 141 and 2 chains (CLICK me for more info!!).
Two alpha (α) subunits and two beta (β) subunits make up up the four polypeptide subunits that make up hemoglobin.
A haemoglobin molecule may carry four oxygen molecules at once because each of the four subunits contains a heme (contains iron) molecule in which the oxygen is attached through a reversible reaction.
Epidermal growth factor receptor
a big, adaptable, multiple-domain protein. involved in the transmission of signals confirmed target for cancer treatment.
-
A sizable extracellular component
-
Transmembrane segment
-
Kinase domain
The transmembrane protein known as the epidermal growth factor receptor (EGFR; ErbB-1; HER1 in humans) is a receptor for extracellular protein ligands that belong to the epidermal growth factor family (EGF family).
Since EGFR was discovered to be an oncogene, "EGFR inhibitors," also known as anticancer therapies, have been created.
These include cetuximab for colon cancer and gefitinib, erlotinib, afatinib, brigatinib, and icotinib for lung cancer. Third generation tyrosine kinase inhibitor Osimertinib was created by AstraZeneca more recently.
Epidermal Growth Factor is a common ligand for EGFR :
-
Released by cells in active growth.
-
Binds EGFR
-
53 amino acid residues forming three disulfide
Extracellular portion bound to EGF
-
[EGF binding induces conformational change
-
Leads to dimerization (quaternary structure)
Transmembrane domain
-
Connects extra- and intra-cellular domains
-
44 residues (alpha-helix)
lntracellular Kinase domain
-
Autophosphorilation, activates signal cascade
-
N-term lobe (mostly B-sheet), C-term lobe (alpha-helices)
---- Summary ----
As of now you know all basics of Fold Structures.
-
Salt bridges.
-
Fold space.
-
Fold structural classification.
-
EGFR, EGF...
-
etc..
________________________________________________________________________________________________________________________________

________________________________________________________________________________________________________________________________