
Molecular Modeling & Drug Discovery
2. Secondary Structure Folding:
Peptide chain
3D-Figure 1. PDB structure of 1TR8 is a archaeal Nascent Polypeptide-associated Complex (aeNAC) with sequence length of 102 and 2 chains (CLICK me for more info!!).
Color identites in polypeptides have specific meaning as indicated from above floating diagram. These notations have been indicated below.
Blue - non polar
Red - polar
Grey - basic
Pink - Alpha carbon
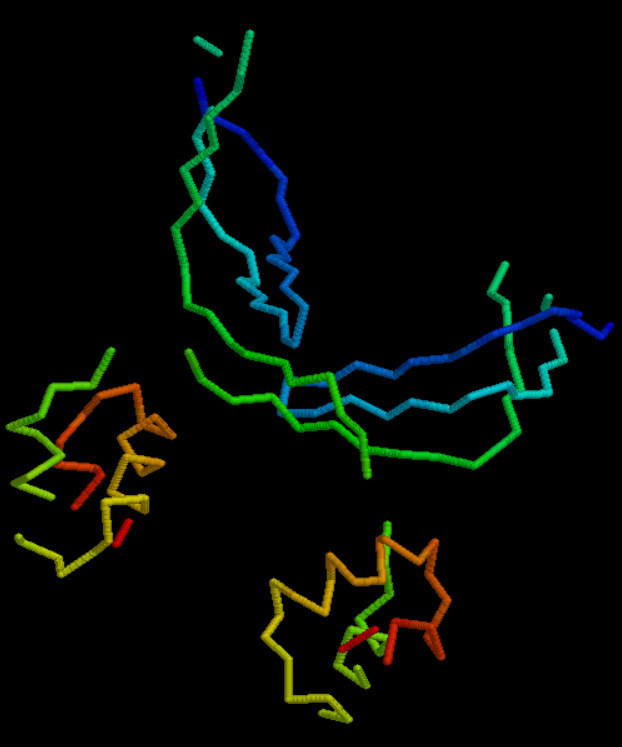
Figure 1. Proteins are held together and given their general form by their protein backbone.
The polypeptide chain's backbone is made up of the peptide bonds connecting the amino acids.
The Ramachandran plot shows the first two torsion angles of the protein backbone, which are designated by Ψ and Φ. Starting from the backbone, the torsion angles in the side chains are indicated by χ1, χ2 etc.
Protein Secondary Structure Elements
Local protein segments are arranged in three dimensions as protein secondary structure.
Alpha helices and Beta sheets are the two secondary structural components that are most frequently found, while beta twists and omega loops also happen. Before three-dimensional tertiary structure, there are intermediate forms called secondary structure elements.
The arrangement of hydrogen bonds between the amino hydrogen and carboxyl oxygen atoms in the peptide backbone serves as the formal definition of secondary structure.
It is also possible to define secondary structure without consideration to hydrogen bonds by looking at the predictable distribution of backbone dihedral angles in a particular area of the Ramachandran plot.
The angle formed by two intersecting planes or half-planes is known as a dihedral angle. It is the clockwise angle between half-planes in chemistry that passes across two sets of three atoms that share two atoms.
.png)
Figure 2. Depiction of 2D dihedral angles in ethane.
The Klyne-Prelog system is used to indicate angles (called either torsional or dihedral angles between substituents around a single bond) between substituents.
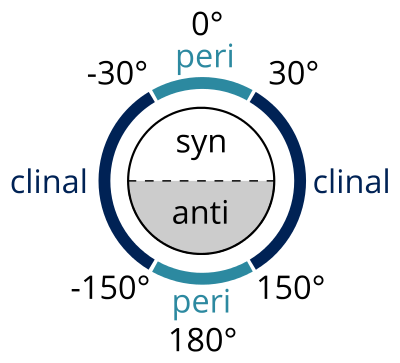
Figure 3. All 2D dihedral angles diagrammatically represented.
syn (s)
torsion angle
:
0° and ± 90°
anti (a)
torsion angle
:
± 90° and 180°
clinal (c)
torsion angle :
:
30° and 150° or –30° and –150°
periplanar (p)
torsion angle
:
0° and ± 30° or ± 150° and 180°
synperiplanar (sp)
torsion angle
:
0° and ± 30°
[syn- or cis- conformation]
synclinal (sc)
torsion angle
:
30° to 90° or –30° to –90°
[gauche or skew]
anticlinal (ac)
torsion angle
:
90° and 150° or –90° and –150°
antiperiplanar (ap)
torsion angle
:
± 150° and 180°
[anti- or trans- conformation]
Torsional strain or "Pitzer strain" refers to resistance to twisting about a bond.

Figure 4. Ramachandran plot for alpha helices and beta sheets depicted with permissible Phi and Psi angles.
A protein's overall three-dimensional arrangement of its polypeptide chain in space is referred to as its tertiary structure.
Internal hydrophobic interactions between nonpolar amino acid side chains and outer polar hydrophilic hydrogen and ionic bond interactions stabilize it most frequently.
3D-Figure 2. PDB structure of 1JKB is a mutant Human Lysozyme (Glu replaced by Ala at position 35) with sequence length of 130(CLICK me for more info!!).
There are numerous ways that the helix, sheet, and loop parts can be combined as a result of interactions between the side chains of the protein's individual amino acid residues.
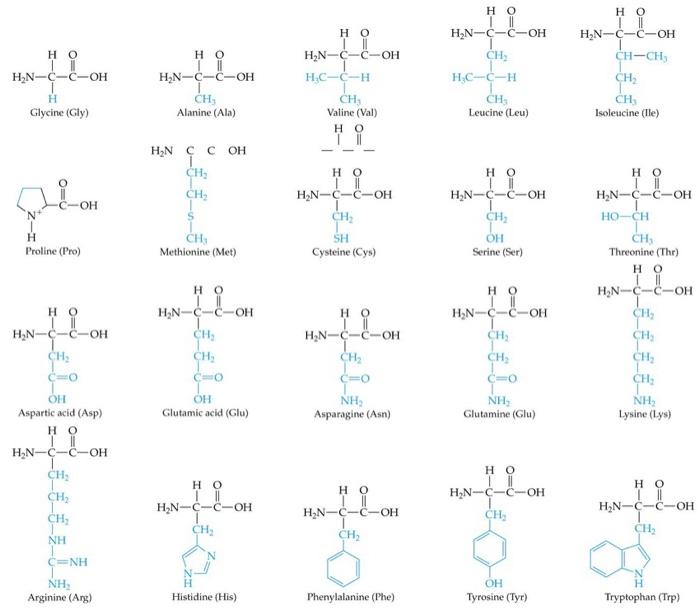
How to remember them all in order, below we have the sequence you have to memorize:
GAVIL
PMCST
AGAGL
AHPTT
As a result, side chains have a far
more significant role in determining the final structure at the level of
tertiary structure.
.png)
Figure 5. Various amino acids and their side chains highlighted in this figure.
Secondary structure is typically produced as a result of backbone interactions (particularly in the case of helices and sheets).
The majority of proteins have a characteristic tertiary structure, which is essential for the protein's correct operation.
Protein folding is the physical process that converts a protein chain to its natural three-dimensional structure, usually a "folded" conformation that allows the protein to operate biologically.
A polypeptide folds from a random coil into its characteristic three-dimensional shape in a rapid and repeatable manner.
Although some portions of functioning proteins may still be in an unfolded state, the right three-dimensional structure is necessary for function, making protein dynamics crucial.
The majority of misfolded proteins are inert because they cannot fold into their original structure, but occasionally they have altered or harmful functions.
Prions, which are infectious forms of misfolded proteins that produce amyloid fibrils, are thought to be the cause of a number of neurodegenerative and other disorders.
3D-Figure 3. PDB structure of 2N53 is a solution structure of ovis aries prion protein with sequence length of 154(CLICK me for more info!!).
PrP proteins with altered structures known as mammalian prions cause contagious, deadly neurodegenerative disorders. Through the development of beta-sheet-rich assemblies that initiate cellular PrP's conformational change, they are self-replicating.
Because the immune system does not develop antibodies for specific protein structures, many allergies are brought on by improper protein folding.
Anfinsen Experiment
Primary protein structure consists of all important information to correctly self assemble back into its functional form.
Anfinsen outlined three requirements:
-
Uniqueness – There is just one minimum free energy conformation for any amino acid sequence.
-
Stability - Lack of reactivity to minute environmental changes due to folding that is most energy-efficient
-
Kinetic accessibility - The activation energy between the various folding processes shouldn't be too high.
Examples of Anfinsen dogma special cases include B. prions and proteins dependent on chaperones to fold correctly.
The thermodynamic theory of Anfinsen states that a protein's lowest potential was corresponding to its native fold energy level.
.png)
Figure 6. An energy map of protein folding has a theorized funnel-like curve called the folding funnel.
A protein chain spontaneously folds during the protein folding process. "Falls" from the unfolded state of higher energy to the one of lower energy folded position.
The native and folded configuration of a protein is at the same time the energetically most advantageous, according to the Anfinsen dogma, which explains why protein folding under physiological conditions typically develops autonomously (without chaperones).
A local minimum in the free energy between potential folds exists in the native form. A model for characterizing the thermodynamics of protein folding is the folding funnel.
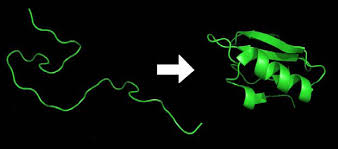
Figure 7. Representation of protein unfolded structure to folded protein structure.
The folding funnel provides an explanation for how a sphere can travel from any point on the perimeter of the funnel to the center.
The folding of a protein would work the same way; regardless of the protein's initial shape, it would simply need to follow the potential gradient in order to achieve its least energy state.
The funnel's widest aperture corresponds to all potential denatured conformations; at this point, the conformational entropy is maximum.
The conformational possibilities decrease as the free enthalpy rises. The semistable intermediates, which are represented by the local minima at the funnel's sides, can help or hinder the creation of the native structure depending on their depth.
The native state, which has a clearly defined shape, is at the bottom.
Levinthal paradox
The Levinthal paradox was coined by Cyrus Levinthal to explain the molecular biology mystery of how an amino acid chain rapidly achieves the functionally folded state of a protein.
Through a thorough examination of all potential conformations, Cyrus Levinthal calculated the time it would take a protein chain to locate its lowest energy conformation.
According to him, a thorough survey of the entire area would require even for chains as short as forty, several times the age of the cosmos. However, it is known that the majority of proteins fold in a few of seconds.
Current protein folding model
-
The "energetic landscape" is funnel-shaped when it folds.
-
The funnel's edges correlate to areas of significant potential energy unfolded states
-
The protein is funneled into lower energy states as it folds till it reaches its original fold, states.
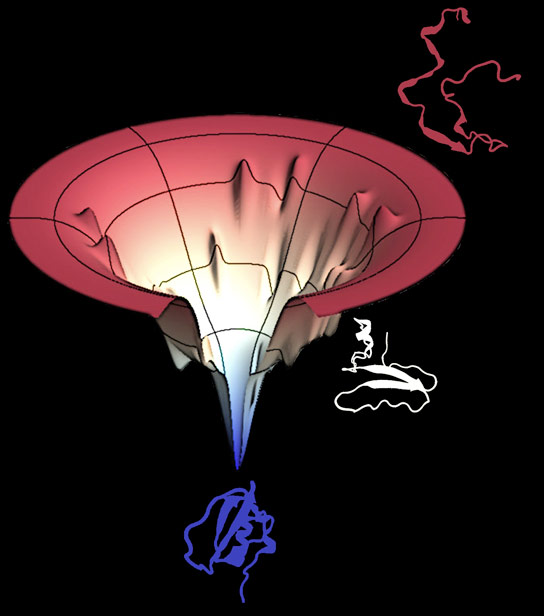
Figure 8. A protein must pass through a series of structures in order to fold into its native state, but parallel processes, ensembles, and dimensions are more important factors in folding.
A golf-course landscape is created by a random searching for the native states, but a rugged pathway deviates from the initial smooth golf-course, creating a directed tunnel where the denatured protein goes through to reach its native structure.
This pathway emphasizes the one-dimensional route of conformation.
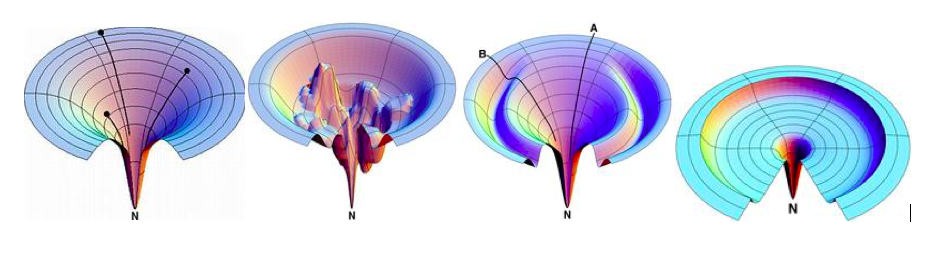
Figure 9. The smooth idealized funnel, the rough funnel, the Moat funnel, and the Champagne Glass funnel are shown in the suggested funnel-shaped energy landscape, from left to right.
-
The Rugged funnel illustrates kinetic traps, energy barriers, and some narrow throughway paths to native state.
-
The Moat landscape illustrates the idea of a variation of routes including an obligatory kinetic trap route that protein chains take to reach their native state.
-
The Champagne Glass landscape involves free energy barriers due to conformational entropy that partly resembles the random golf-course pathway in which a protein chain configuration is lost and has to spend time searching for the path downhill.
The Idealized smooth funnel is the best option. The rugged funnel is the next best option, but has a higher potential for failure. The Moat funnel is the worst option, as it has the highest potential for failure and the least potential for success. The Champagne Glass funnel is the least bad option, as it has the lowest potential for failure and the highest potential for success.
Small natural proteins folding
-
It appears that the protein sequences have evolved to have smooth energy landscapes devoid of notable dips in addition to forming their native shapes.
-
Highly "cooperative" protein folding has been seen in small naturally evolving proteins.
-
Once the protein starts folding, the transition from the unfolded to the folded form happens very quickly, making it impossible to distinguish distinct intermediate structures.
Large protein folding
-
The mechanisms that underlie the folding of larger proteins (those with more than 100 residues) seem to be similar.
-
More modular folding pattern - Partially folded proteins may interact inappropriately, resulting in the development of toxic aggregates.
-
Higher complexity slows down folding.
-
Transition stages on the road to the native state can be segregated.
-
Molecular chaperones are proteins that aid in the proper folding and prevent aggregation of other proteins (e.g. Hsp90)
---- Summary ----
As of now you know all basics of Secondary Structures.
-
Torsion angles.
-
Anfinsen experiment.
-
Peptide bonds.
-
Current protein folding model.
-
etc..
________________________________________________________________________________________________________________________________

________________________________________________________________________________________________________________________________